“What we lack is not hope, but time to wait for hope.”
Writing |Ling Jun
Source | “Medical Community” Public Account
On September 22, 2022, the New England Journal of Medicine published results of a phase 3 clinical trial showing that an innovative genetic drug, tofersen, slowed or even reversed some ALS The patient’s disease progression.
“I can walk around the house without my crutches and stop taking some painkillers.” Les Wood, 68, was the first participant in the trial, and has now suffered from ALS10 year.

Walking alone at home after Les Wood treatment
ALS, or amyotrophic lateral sclerosis (ALS), is best known for being afflicted by physicist Stephen Hawking. Hawking was diagnosed at the age of 21 and fought the disease for 55 years before his death. But he is a special case. The average survival time of patients with ALS is only 3 to 4 years, and less than 10% can live for more than 10 years.
After being diagnosed with ALS in 2012, Les Wood lost the ability to work, along with his nurse wife. As a degenerative neurological disease, people suffering from ALS will gradually shrink the muscles of the whole body, lose the ability to move independently, and later have difficulty swallowing, until respiratory failure and death.
A healthy person is like being frozen into ice by time. Some statistics suggest that there are approximately 500,000 ALS patients worldwide.

Stephen Hawking
At present, the etiology and pathogenesis of ALS is unknown, but about 2% of patients are thought to be caused by nerve damage caused by mutations in the SOD1 gene that produce a toxic protein. And Les Wood is exactly that 2%.
In 2016, as one of the first patients enrolled, Les Wood participated in the tofersen clinical trial in a wheelchair. The researchers recruited a total of 108 patients with ALS with mutations in the SOD1 gene. The drug group underwent a monthly lumbar puncture, in which a needle is passed between the bones of the spine and the drug is injected directly into the spinal fluid.
The initial 6-month trial was not very good. There were no clinically significant differences in related disease scores, mobility, and lung function between the drug and placebo groups.
But the trial also found that compared to the placebo group, the total concentration of SOD1 protein in the cerebrospinal fluid of the patients in the drug group was significantly lower. Based on this promising sign, the researchers extended the dosing cycle — 63 patients in the drug group continued the drug for 12 months, while another 32 in the placebo group started the drug from scratch.
For patients with “slow disease progression” prior to treatment, muscle strength improved after 1 year of treatment and overall disease severity remained stable at final assessment. For patients with “more severe disease”, the rate of muscle function decline was also significantly slower.
At the same time, the earlier the drug is administered, the more effective the treatment will be. In addition to Les Wood, another patient who received treatment said that he was able to write greeting cards again.
Compared to the negative trial results at 6 months, the researchers analyzed the reason why the efficacy was shown after 1 year, “the lag is only a reflection of motor neuron healing and clinical manifestations. The time required for the difference.”
Uncertain efficacy and patient ‘life-saving straws’
In the past 40 years of research, only two drugs have been approved by the US FDA for the treatment of ALS in the world, but they are incurable and can only delay ALS to a very limited extent. Frozen disease course. One of the drugs, relevant data shows, may only delay survival by 2-3 months.
Earlier, another new drug for the treatment of ALS, AMX0035, aroused heated discussions, and its 137-person clinical trial showed:
It is safe and can still show a certain survival benefit after about 3 years of use. It reduced the risk of death by about 44% compared to a placebo. But AMX0035 found no statistically significant difference in survival during the 6-month randomization period.
But in March, the FDA’s Peripheral and Central Nervous System Drugs Advisory Committee concluded that the study failed to provide “sufficient evidence,” including a small trial size and missing data. The drug was initially rejected.
Industry analysis, perhaps because of Aduhelm’s lessons learned – Aduhelm was approved by the US FDA for the treatment of Alzheimer’s disease on June 7, 2021, but due to the lack of credible efficacy data , making the FDA questionable.
Patients’ attitudes are diametrically opposed to those of experts. After the FDA took a negative attitude towards the listing of AMX0035, according to overseas media reports, thousands of emails were sent to the FDAOffice of the Commissioner, they asked the FDA to reconsider the approval of the drug. This month, the same committee met again, this time recommending approval of AMX0035.
Similarly, compared to most blockbuster innovative drugs in other disease areas, tofersen’s role is very limited, said Dr. López de Munain of the Spanish Institute of Neurodegenerative Diseases, “improving biological Markers are one thing, recovery or prevention of disease is another.”
In a way, the root cause of tofersen’s attention is that ALS patients try to grab any hopeful “life-saving straws”, no matter what the outcome, they have no choice .
Stop partial ALS at its source
Tofersen is an antisense oligonucleotide (ASO) drug.
As one of the representatives of small nucleic acid drugs, the mechanism of action of ASO drugs is generally understood in principle. Through the principle of base pairing, it binds target mRNA and inhibits gene expression to prevent “bad” proteins. produce. The advantage is that in theory, as long as the disease-causing gene is discovered, the corresponding drugs can be specifically designed to suppress the disease from the source.
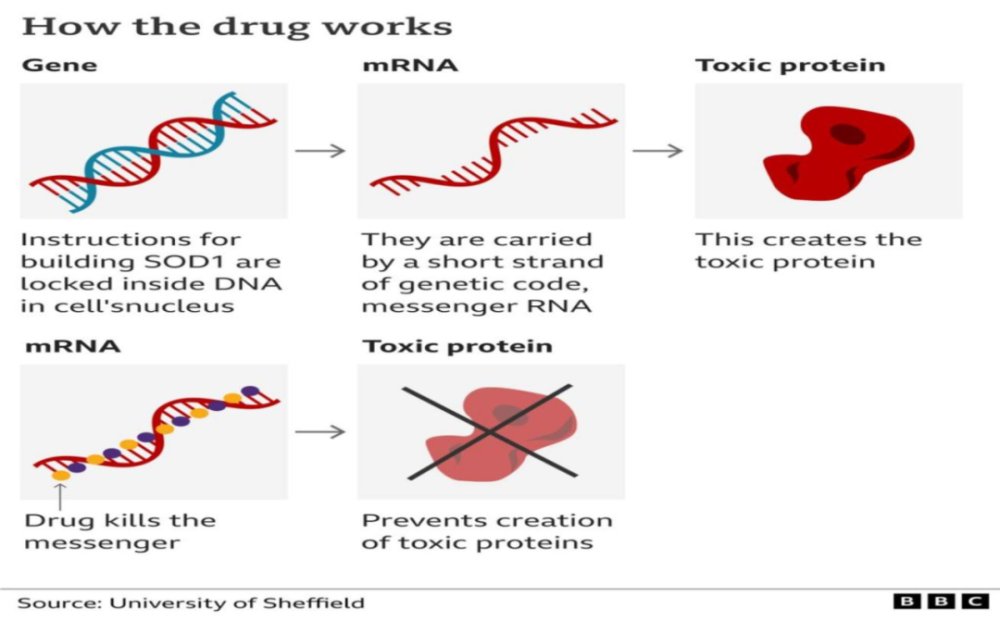
In addition to tofersen, in a study published in Nature Medicine in December 2021, scientists also designed another ASO therapy for ALS, targeting the target gene C9orf72— – Compared with SOD1 gene mutation, which is more common, a patient’s ALS functional score and other indicators were basically stable or slightly improved after treatment.
But for ALS, the shortcomings of related therapies are also very obvious. The first is the limitation of “target genes”.
Among people with ALS worldwide, one data suggests that only about 20% of cases may be related to genetics and genetic defects. For this tofersen, it only applies to ALS patients with mutations in the SOD1 gene—about 2 percent.
On the other hand, although tofersen has been called a “landmark breakthrough” in slowing the course of ALS, it still doesn’t lead to a complete cure. Early in the disease, the drug prevented further nerve damage and failed to generate new motor neurons. It can take up to a year for partially damaged neurons to recover and form new connections with muscle tissue.
In addition, clinical trial results underscore that tofersen should be used early, but longer-term efficacy and safety data remain to be validated.
But co-author of the clinical study, Professor of Neurology at the University of Sheffield, Dame Pamela Shaw, believes that what makes ALS so terrifying is the speed of change, which, if it stabilises the disease the most, is A huge achievement. At the same time, if the drug is finally proved successful, it will also help to accelerate the research progress of similar methods of gene drugs, and establish a breakthrough for the treatment of ALS from the mechanism.
It can be seen that, from traditional chemical drugs and protein drugs to today’s hot gene/cell therapy drugs, more and more “terminal illnesses” have the possibility of being cured. Recently, scientists have also developed an ALS therapy based on genetic engineering combined with stem cell therapy. The results of the Phase 1/2a clinical trial were published in Nature Medicine on September 5.
But as a family member of a child with a rare disease previously told the “medical community”, “What we lack is not hope, but the time to wait for hope”, for ALS patients, The average lifespan for them to wait is less than 5 years.
It is reported that the US FDA has accepted tofersen’s marketing application in July this year, and included it in the priority review qualification, and it is expected to make a ruling on January 25 next year.
References:
[1]’Truly remarkable’drug helps motor neurone disease, https:https://www.bbc.com/news/health-62851186
[2]FDA seems poised to approve a new drug for ALS, but does it work?, https:https://www.gpb.org/news/shots-health-news/ 2022/09/22/fda-seems-poised-approve-new-drug-for-als-does-it-work
[3]Biogen trumpets data from ALS trial as FDA decision looms, https://pharmaphorum.com/news/biogen-trumpets-data-from-als-trial-as- fda-decision-looms/
[4]Experimental drug shows signs of slowing motor neurone disease, https://www.theguardian.com/society/2022/sep/21/experimental-drug-slowing-motor -neurone-disease
[5] FDA Committee, in Reversal, Favors AMX0035 Approval for ALS, https://alsnewstoday.com/news/fda-committee-reversal-favors-amx0035-approval-als /
[6]How far is the spring of graduating people? AlbThe land of blood and honey behind rioza, https:https://baijiahao.baidu.com/s?id=1738010653107880166&wfr=spider&for=pc
Source: Medicine
Proofreading: Zang Hengjia
Editor in charge: Tian Dongliang
* The medical community strives for the accuracy and reliability of its published content when it is reviewed and approved, but does not regard the timeliness of the published content, and the accuracy and completeness of the cited materials (if any), etc. Make any promises and guarantees, and do not assume any responsibility for the outdated content, possible inaccuracy or incompleteness of the cited information. Relevant parties are requested to check separately when adopting or using it as a basis for decision-making.