https://
There is a group of people with slender limbs, excellent height, and excellent performance in some aspects, which can be described as “genius”, but as everyone knows, the gift of fate is always withdrawn early. So, who is the culprit?
In 1896, French paediatrician Antoine Marfan first reported a case of disproportionately slender and long limbs, fingers, and toes (spider fingers/toes), poor muscle development, and a combined spinal side Bent 5-year-old female patient.
Follow-up studies have found that this is a clinical syndrome, and patients can also show multi-system manifestations such as ocular and cardiovascular abnormalities. “Marfan syndrome (MFS, also known as spider disease, genius disease) )” hence the name.
Behind talent is cruel destiny
One,
Pathogenesis
MFS is a rare genetic disease. The mutation of the FBN1 gene on chromosome 15 leads to abnormal expression of microfibrin-1, an important component of the extracellular matrix, and the lesions invade the connective tissues of the whole body. It mainly affects the eye, bone, and cardiovascular system [1].
Patients often die suddenly due to cardiovascular complications such as aneurysm rupture, arterial dissection, and heart failure. Untreated patients generally live less than 40 years old.
According to reports, the prevalence of KFS is about 6.5/100,000 people, and many “giant” athletes such as Ver-Hyman, Zhang Jiadi, and Huo Xuan were killed by the disease.
Second,
Clinical manifestations
Patients have complex phenotypes with large clinical variability. Even among different patients in the same family, their phenotypes vary widely. According to literature reports, patients mainly have the following three characteristics[2-3]:
1►Eye performance
Mainly including lens dislocation, high myopia, retinal detachment, glaucoma, strabismus and amblyopia, etc. Other rare manifestations include sunken eye, congenital ptosis, miosis, premature cataract, pigment membrane inflammation and iris tremor.
2►Bone representation
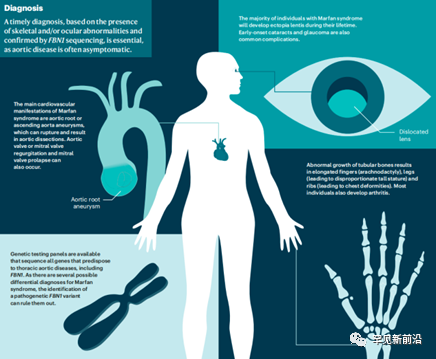
A long head, narrow face, raised palate, and elongated limbs (especially the fingers and toes) are characteristic of the disease. The patient’s slender limbs, excellent physique, and arm span-to-height ratio >1.03 have diagnostic value.
Before skeletal maturity, patients may develop kyphosis or scoliosis due to sternal depression/bulge (chicken breast, pectus excavatum), vertebral deformities, and sometimes joint laxity or recurrent joint dislocations . Spider fingers (toes), thumb sign, and finger-wrist sign are also important clinical signs.
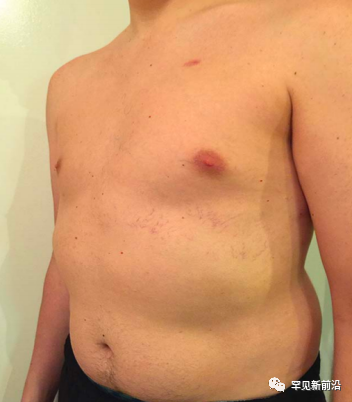
Figure 1 Chicken Breast
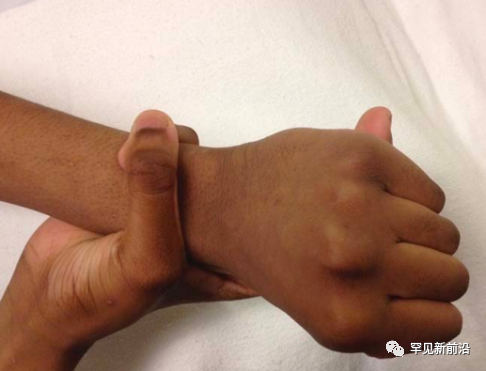
Figure 2 Wrist sign
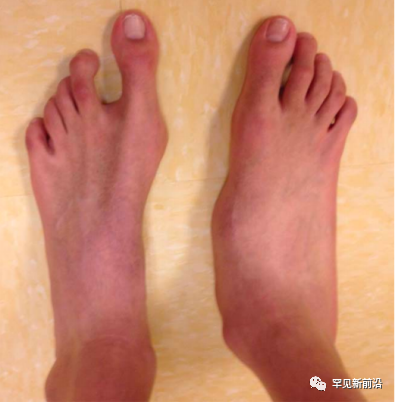
Figure 3 Foot deformities
3 ►Cardiovascular abnormalities
is the most dangerous lesion of the disease and the leading cause of death. Aortic root dilatation, ascending aorta dilatation, aortic dissection and heart failure and other serious cardiovascular diseases can lead to sudden death of patients. Other manifestations include heart valve insufficiency, heart failure, and various arrhythmias, such as conduction block, atrial fibrillation, and ventricular arrhythmias.
In addition, KFS patients mayOther system performance [4]:
1.Respiratory system: Mainly spontaneous pneumothorax, which can recur due to rupture of apical bullae;
2.Genitourinary organs: polycystic kidney disease, female uterine prolapse, dysuria;
3.Nervous system: Dura (spinal) membrane burst, nerve root irritation symptoms;
4. Muscle:Muscular dysplasia;
5.Body surface part: dermatoglyphic changes, hernia, etc.;
Three,
MFS Diagnostics
According to the latest “Revised Ghent Criteria (2010)”, the diagnostic requirements for MFS are as follows [5]:
1. If there is no family history of MFS, either of the following:
1) Aortic root aneurysm or aortic root dissection (Z≥2) and lens dislocation
2) Aortic root aneurysm or aortic root dissection (Z≥2) and FBN1 mutation
3) Aortic root aneurysm or aortic root dissection (Z≥2) and systemic score≥7
4) Coexistence of lens dislocation, FBN1 mutation and known aortic root aneurysm or aortic root clip
Layer
2. If there is a family history of MFS, either of the following:
1) Lens dislocation
2) Whole body system score≥7
3) Aortic root aneurysm or aortic root dissection (Z≥2 for those over 20 years old, Z≥3 for those under 20 years old).
Note: The total score of the whole body system is 20 points, and the score ≥ 7 points indicates that the whole body system is involved;
Fourth,
Can MFS be prevented?
MFS is an autosomal dominant disorder. To try to avoid the birth of affected offspring, patients should be provided with genetic counseling to inform their offspring that the genetic risk is 50%. In addition, prenatal diagnosis of high-risk pregnancies can be performed with ultrasound during pregnancy.
For individuals suspected of MFS, early diagnosis and early intervention should be sought. There is no specific treatment for this disease, and symptomatic treatment is still the mainstay, including drug therapy based on beta-blockers and surgical treatment based on vascular replacement and valve repair [6]. But overall, the treatment effect of this disease is not satisfactory, and the prognosis of patients is extremely poor.
References
1. Lucia Brunello, Laura Marshall. Marfan syndrome. Rev Dis Primers.2021 Sep 2;7(1):63. doi: 10.1038/s41572-021-00304-y.
p>
2. Chu Gejin, Zhang Qingjiong, Guo Xiangming. Advances in clinical diagnosis and molecular biology of Marfan syndrome. Chinese Journal of Practical Ophthalmology, 2002, 20(4), 245-249.
3. Adam DB, Paul DS. Marfan Syndrome: A Clinical Update. J Am Acad Orthop Surg, 2017;25(9):603-609. doi: 10.5435/JAAOS-D- 16-00143.
4. Reed E Pyeritz. Marfan syndrome: improved clinical history results in expanded natural history. Genet Med, 2019, 21(8):1683-1690. doi: 10.1038/s41436-018- 0399-4.
5. Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010, 47(7): 476-485. doi: 10.1136 /jmg.2009.072785.
6. Yunxiao Yang, Xiubin Yang. Research progress of vascular smooth muscle cell phenotype conversion and vascular lesions in Marfan syndrome. Chinese Journal of Cardiovascular Diseases, 2022, 50(4): 410- 414.