This article was written by carollero
Dean’s face looks like a “sad bun.”
As a child, Dean’s face began to develop open wounds that would not heal for a long time.
This wound is like an erupting volcano, and the blood-colored magma starts from the middle of the face and spreads to the sides. In the end, only a small patch of normal skin tissue remained on Dean’s face.


Young Dean (Credit: The Feed SBS)
Since then, Dean’s face has “rotten” like the rest of the skin on his body.
Crisp like a butterfly wing, it “breaks” when touched
This strange skin condition started many years ago.
When Dean was 12 hours into the world as a newborn, tiny pink blisters began to appear on his skin. The blisters were plump and transparent, and were mainly concentrated on Dean’s buttocks.
At the time, Dean’s parents thought it was a common baby allergy and didn’t take it seriously.
The doctor who delivered Dean was worried. He recalled his previous delivery experience, and another baby who had encountered a similar situation with Dean came to his mind.
He clearly remembers that the baby was also full of these blisters shortly after birth, which ruptured with light friction and caused great pain in the baby. , he always cried repeatedly.
Finally, the baby died after 10 days.
Therefore, the doctor warned Dean’s parents to pay close attention to the development of these blisters. “This is a serious disease.”
In the years that followed, the disease showed its fangs and claws as expected.
New blisters continued to appear on Dean’s skin. These transparent blisters were even flaky, and from a distance it looked like a layer of “new skin” had grown.
But this “new skin” is separated from Dean’s body. Dean’s skin is like a fragile butterfly wing, which will easily break as long as it is rubbed or injured. Ulcerated water, extremely painful.
At the worst, when Dean and his parents went to the supermarket, they would receive everyone’s attention, and children of the same age were always very frightened. Because only a little of normal skin remains on Dean’s face, “the whole face is gone.”
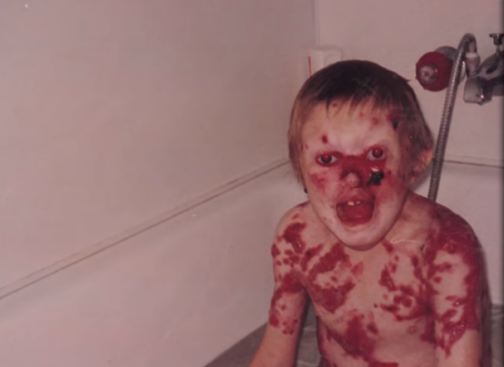
Young Dean (Credit: The Feed SBS)
In this way, Dean’s skin experienced repeated ruptures and slow regeneration. Even as an adult, he still needs to spend three and a half hours a day carefully cleaning his body and wrapping a clean bandage on himself.
“My skin is very fragile every time I need to grab something. I’m an adult and it still takes a lot of energy and time to do it A basic sandwich breakfast.”

Dean’s daily “work” (Photo: The Feed SBS)
However, despite the hardships of Dean’s life, the doctor sighed. BecauseFor the fact that most patients with the disease do not survive infancy, Dean’s survival has been a miracle.
The culprit
Dean suffers from a disease called epidermolysis bullosa (EB), a rare genetic disorder with 16 genes that have been found to be involved in the disease. Mutations in at least one of these genes cause epidermolysis.
Some scholars have interpreted this. Human skin consists of three layers: from outside to inside, it is divided into epidermis, dermis and subcutaneous tissue. In people with healthy skin, there are protein anchors at the epidermis-dermis junction that prevent cells from moving against each other.
But in EB patients, the lack of protein anchors that hold the skin together results in very fragile skin. Even mild mechanical friction (such as friction or pressure) or trauma can separate the skin layers, creating blisters and painful wounds.
Dean’s outer skin is due to non-functional laminin, the cells in the epidermis are fragile and easily damaged, so friction or small trauma can cause interlayers of the skin Separate, form blisters.
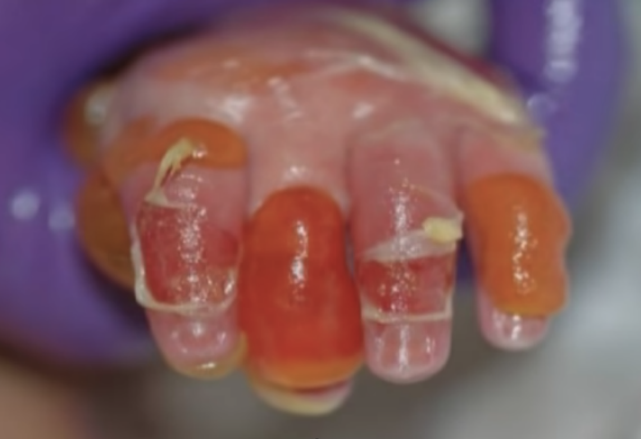
Blister ruptured wound (Source: YouTube)
It is estimated that EB is diagnosed in 20 cases per million live births and 9 cases per million population. Of these cases, approximately 92% had epidermolysis bullosa simplex (EBS), 1% had junctional epidermolysis bullosa (JEB), and 5% had dystrophic epidermolysis bullosa Symptomatic (DEB) and 2% Unclassified.
Of the first two types, the most severe form is generalized severe form of junctional epidermolytic bullosa (formerly known as Herlitz JEB), which has systemic symptoms and Signs are very serious.
From birth or early infancy, children with JEB generalized severe disease develop blisters on most areas of the body.The blisters can also affect mucous membranes, such as the mouth and digestive tract This can greatly increase the difficulty of eating and digesting food, leading to malnutrition and slow growth in many children.
These extensive blisters rupture and regenerate on the surface, often leading to the formation of granulation tissue, which is extremely easy to erode and bleed, increasing the risk of shock and infection. At the same time, the accumulation of granulation tissue in the airways can also make breathing difficult.
A 2014 retrospective study concluded that despite a clear diagnosis, children with JEB die in infancy.
Breaking the game: finding a way from the source
Clinically, it is relatively easy to diagnose EB through blood samples and genetic testing, or by direct needle biopsy at the wound edge using immunofluorescence localization. However, the treatment of EB is a difficult problem.
Doctors are always trying new treatments.
As of 2008, clinical studies at the University of Minnesota have explored allogeneic stem cell transplantation for borderline EB, which was used to treat a 2-year-old and his older brother.
In addition to these two patients, the researchers plan to conduct clinical trials on 30 remaining subjects. But they found that the immune cell depletion required before stem cell transplantation can lead to a serious risk of infection, leading to massive blisters and skin erosions. Of the only group of subjects, at least four died in preparation for stem cell transplantation.
Stem cell transplants don’t work, so “make a skin”—transplant skin derived from genetically modified stem cells to the surface of the wound.
Heisen is a Syrian child with EB. 80% of his epidermis is exfoliated, leaving the dermis exposed. He needs to wear a clean bandage every day like Dean to reduce contact. with infection.
Local scientists chose to use genetically modified stem cells to regenerate his entire human epidermis. They first removed a piece of Hessen’s skin and cultured it in a medium, then used gene editing technology to change a specific mutant gene to a normal gene, and then cultivated this part of the normal skin and transplanted it to Hessian’s body.
Eventually, Hesson becomes a normal boy.

Researchers created this skin from genetically modified cells (Credit: CMR unimore)
However, not all children can become Hessians.
Hessian’s diagnosis was a rare JEB, a type of EB with well-defined sites of genetic mutations, which were mapped to LAMA3, LAMB3, LAMC2, and COL17A1.
The treatment of other types of EB is still groping in the fog.
Recent research has focused on altering the mix of keratin proteins produced in the skin. There are 54 known keratin genes that determine keratin synthesis, most of which have similar basic structure and function but differentiate into different cells under different conditions.
Scientists therefore believe that by controlling the conditions, transfer of the mutated, dysfunctional keratin gene to the fully functional keratin gene can reduce symptoms in children >.
For example, sulforaphane, found in broccoli, is injected into pregnant mice (5 µmol/day = 0.9 mg) and topically applied to newborn mice (1 µmol/day = 0.9 mg) 0.2 mg), the blisters in the mouse model were reduced to the point of being unrecognizable.
However, since these studies have not yet been applied to the clinic, there still seems to be no clear cure for the disease EB. Current disease management focuses on wound care, pain control, infection control, nutritional support, and prevention and treatment of complications.
The Butterfly Needs Pampering
At the same time, researchers believe that the psychological needs of these children also need to be addressed.
On this basis, a survey of 11 families affected by the disease in The Journal of Clinical and Aesthetic Dermatology suggests that both the public and health care providers are aware of the disease Inadequate, so there is always some insensitive, unfriendly evaluation of adults and bullying among children in society.
The educational film produced by the Dateln Children’s and Adolescent Hospital in Germany points out that the care of children with EB requires not only attention to hinder the development of chronic pain, but also the importance of families, doctors, and psychologists. Because the mental health of these children also cannot be ignored.

Lonely Dean (Source: The Feed SBS)
Dean, who has grown up to now, is such a patient group that needs attention.
When Dean finally began to slowly integrate into the society, he said, “I hope I can find a sincere relationship like everyone else in the future.” (Content review: gyouza)< /p>
This article has been professionally reviewed by Dr. Chen Jiaqi, Chief Physician, Department of Dermatology, The Second Affiliated Hospital of Zhejiang University School of Medicine
Image source: The Feed SBS
References (swipe up to view):
1. Bardhan, Ajoy; Bruckner-Tuderman, Leena; Chapple, Iain L. C.; Fine, Jo-David; Harper, Natasha; Has, Cristina; Magin, Thomas M.; Marinkovich, M . Peter; Marshall, John F.; McGrath, John A.; Mellerio, Jemima E. (2020-09-24). “Epidermolysis bullosa”. Nature Reviews Disease Primers. 6 (1): 1–27. doi:10.1038 /s41572-020-0210-0. ISSN 2056-676X.
2. Hon, Kam Lun Ellis; Li, Joshua J.; Cheng, Bernadette L.; Luk, David C.; Murrell, Dedee F.; Choi, Paul C. L.; Leung, Alexander K. C. (2015-03-04). “Age and etiology of childhood epidermolysis bullosa mortality”. Journal of Dermatological Treatment. 26 (2): 178–182. doi:10.3109/09546634.2014.915002. ISSN 0954-6634. PMID 24724596. S2CID 33722635.
3.”Epidermolysis Bullosa Clinic Frequently Asked Questions”. Stanford Medicine — Dermatology. Retrieved 13 April 2018.
4. James, William; Berger, Timothy; Elston, Dirk (2005). Andrews’ Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216- 2921-0.
5. Freedberg, et al. (2003). Fitzpatrick’s Dermatology in General Medicine. (6th ed.). McGraw-Hill. ISBN 0-07-138076-0.
6. Fine, Joe-David; Manes, Becky; Frangoul, Haydar (July 2015). “Systemic granulocyte colony-stimulating factor (G-CSF) enhances wound healing in dystrophic epidermolysis bullosa (DEB): Results of a pilot trial”. Journal of the American Academy of Dermatology. 73 (1): 56–61. doi:10.1016/j.jad.2015.04.015. ISSN 0190-9622. PMID 25956659.
7.Hirsch, T; Rothoeft, T; Teig, N; Bauer , JW ; Pellegrini, G; De Rosa, L; Scaglione, D; Reichelt , J ; Klausegger , A ; Kneisz , D ; Romano, O; Second Second, A; Contin , R ; Enzo, E; Jurman , I ; Carulli , S ; Jacobsen , F ; Luecke , T ; Lehnhardt , M ; Fischer , M ; Kueckelhaus , M ; Quaglino, D; Morgante, M; Bicciato, S; Bondanza, S; De Luca, M (16 November 2017). “Regeneration of the entire human epidermis using transgenic stem cells”. Nature. 551(7680):327–332. Bibcode:2017Nature.551..327H. doi:10.1038/nature24487. PMC 6283270. PMID 29144448.
8.Kerns, Michelle L.; DePianto, Daryle; Dinkova-Kostova, Albena T.; Talalay, Paul; Coulombe, Peter A. (2007-09-04). “Reprogramming of keratin biosynthesis by sulforaphane restores skin integrity in epidermolysis bullosa simplex”. Proceedings of the National Academy of Sciences. 104(36): 14460–14465. Bibcode:2007PNAS..10414460K. doi:10.1073/pnas.0706486104. ISSN 0027-8424. PMC 1964870. PMID 17724334.
9.https:https://www.livescience.com/60897-boy-gets-new-skin-gene-therapy.html