“Every child is born with a message that God is not disappointed with mankind.” – Tagore
It is said that every child is a gift from God, but there are such little “angels” who are born with the same appearance as other babies, but their faces are always filled with splendid expressions. The smile, as everyone knows, is a joke by fate…
Your smile hurts
In 1965, British paediatrician Harry Angelman first reported three mentally retarded, always smiling, unable to speak patients, who looked like a child in a painting in the Castelvecchio Museum who was proud of completing a puppet smile, so it is called “happy puppet disease”.
Subsequent research found that this is a rare, hereditary, neurodevelopmental syndrome, and in honor of it, “Angelmansyndrome (AS)” was named.

Children with “Angel Syndrome”[1]Smile in oil painting
One. Pathogenesis
AS is a neurodevelopmental disorder caused by abnormal expression or functional defect of the UBE3A gene in the maternal 15q11-13 chromosomal region. The prevalence in European and American populations ranges from 1/24,000 to 1/12,000. Most of them are sporadic in my country, and there is no relevant epidemiological investigation report.
The patient’s nervous system, mainly the abnormal ubiquitination of the substantia nigra, striatum, hippocampus and cerebellar Purkinje cells, in the fetal period and neonatal period, the clinical phenotypes are mostly It is normal and typically presents with typical AS symptoms after 1 year of age [2].
II. Clinical manifestations
With reference to the 2005 version of the consensus on clinical diagnostic criteria, the clinical features of AS can be divided into [3,4] according to the frequency of occurrence:

1, all present(100% of patients)
(1) Developmental delay: developmental milestones are delayed without regression;
(2) Movement or balance disorders: usually ataxia and/or limb tremor;
(3) Language barriers: no or very little vocabulary, repetitive language and non-verbal communication skills are stronger than expressive language skills;
(4) Abnormal behavioral characteristics: frequent laughing or smiling, obvious excited or happy behavior, often accompanied by clapping or hyperactivity;
(5) Normal maternal history and birth head circumference, no birth defects and abnormal biochemical indicators.
2. Frequent manifestations(>80% of patients)
(1) Small head circumference or slow growth (
(2) Seizures (before age 3);
(3) Abnormal EEG: characteristic high-amplitude spike-slow wave.
3. Correlation performance(20%-80% of patients)
Occipital flattening, tongue sticking out, salivation, feeding difficulties in infancy, hypotonia, excessive chewing action, strabismus, large mouth and wide teeth, hypopigmented skin compared to family members (only seen in absent), lower extremity tendon hyperreflexia, fondness for upper extremity lift when walking, increased thermal sensitivity, wide footbed, ankle varus or valgus, poor sleep, obesity (more common in older, nondeficient patients), scoliosis Bending, constipation, etc.
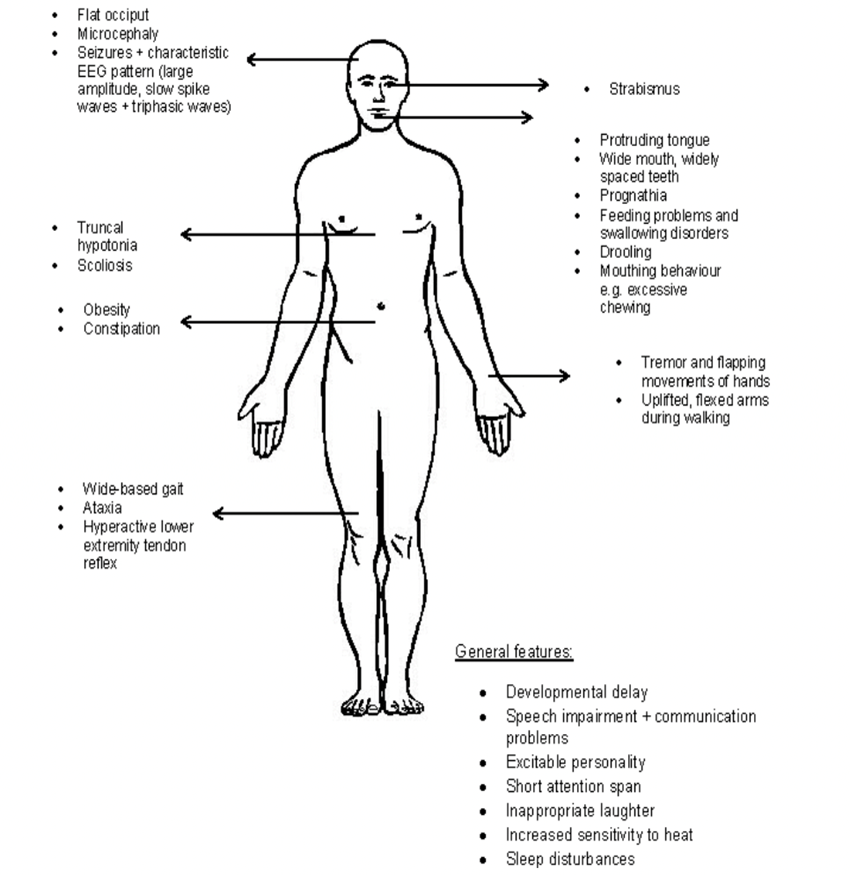
Clinical features of AS[4]
Three. Auxiliary examination
1. Genetic testing
for the diagnosis of the disease. It mainly includes DNA methylation analysis (methylation multiplex ligation-dependent probe amplification technology/MS-MLPA, methylation PCR) and UBE3A gene sequence analysis. In addition, SNP-array analysis can detect microdeletion of chromosome 15 and paternal uniparental disomy.
2. EEG
It is helpful for early diagnosis of the disease before the patient has obvious clinical symptoms or genetic diagnosis. The currently recognized EEG features of AS include: ① δ pattern; ② θ pattern; ③ posterior head spikes and slow waves.
IV. Diagnosis
A diagnosis can be made when the consensus of clinical diagnostic criteria for AS and/or molecular genetic testing indicate that the maternal UBE3A allele is defective in expression or function.
Patients with different pathogenesis adopt different diagnostic methods during the diagnosis process. DNA methylation analysis is the first choice for genetic testing, which can diagnose AS caused by maternal 15q11.2-q13 deletion, paternal uniparental disomy, or imprinting defects, accounting for about 80% of all patients.
Sequencing of the UBE3A gene can be performed in individuals with normal DNA methylation analysis, which can lead to the diagnosis of AS in approximately 10% of patients. In addition, there are still about 10% of patients who cannot achieve molecular genetic diagnosis due to the unclear pathogenic mechanism, and can only make clinical diagnosis based on typical manifestations.
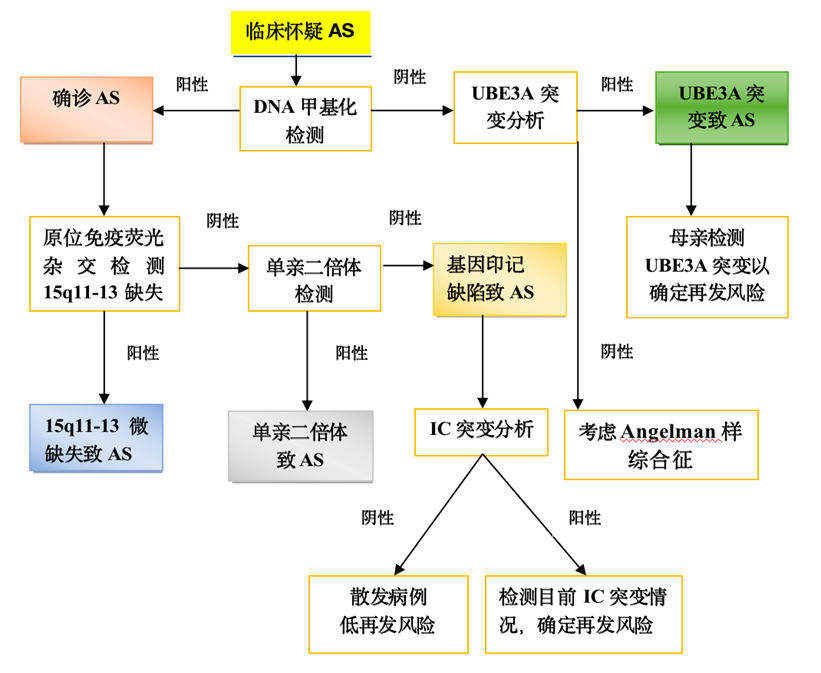
AS diagnosis and treatment flow chart[2]
V. Treatment
As of now, there is no specific treatment for AS, and the main work is health management after diagnosis. At present, active symptomatic and supportive treatment is mainly aimed at clinical manifestations such as feeding difficulties, epilepsy, abnormal behavior, speech disorders, and scoliosis, so as to improve the quality of life of children.
Molecular targeted therapy is the current research hotspot for AS treatment and the only possible cure.
A number of gene therapies are being actively pursued, such as activation of methylation-silenced paternal UBE3A alleles using telomerase inhibitors or antisense oligonucleotides. In order to achieve the purpose of increasing the expression of the paternal UBE3A allele, complementing the maternal UBE3A allele defect, and finally treating the disease.