Patient General:
Female, 42, housewife
Main symptoms and appeals: cough for one year, sputum and blood for half a month. History of present illness:
The patient developed cough, white sputum, no obvious fever and chills 1 year ago without obvious incentive, and felt chest tightness and shortness of breath after exercise. The symptoms became obvious when the weather changed, and the patient went to the local hospital. The patient was treated with anti-infective and phlegm-relieving treatment. The patient’s symptoms did not improve significantly, and there was no significant change in the re-examination of lung CT. The patient went to the doctor in January this year, considering “interstitial lung disease?”, and was treated with Fulusi anti-fibrosis, Asmemet cough and Quanzaile inhalation, and the patient still had cough symptoms. Half a month ago, the patient had symptoms of blood in sputum, and there was one mouthful of blood in sputum every morning. The patient went to the outpatient department of our hospital for further treatment and was treated with cefixime and erdosteine. After further treatment, he was admitted to the hospital with “interstitial lung disease”.
Past and personal history:
The patient complained of low vision in both eyes, yellow-white eyelashes and eyebrows, and light hair color since birth, and has not been diagnosed and treated.
History of postpartum hemorrhage.
Denying alcohol and tobacco addiction.
Outside hospital inspection:
Chest CT report on 2021-12-18 from a traditional Chinese medicine hospital in a district of Shanghai: chronic interstitial lung disease and fibrosis with bilateral pleural thickening and adhesion in both lungs; small nodules in both lungs (located in the upper right , lower left, the larger diameter is 5.5mm, the boundary is clear); the soft tissue density shadow of the anterior superior mediastinum (about 17*9mm).
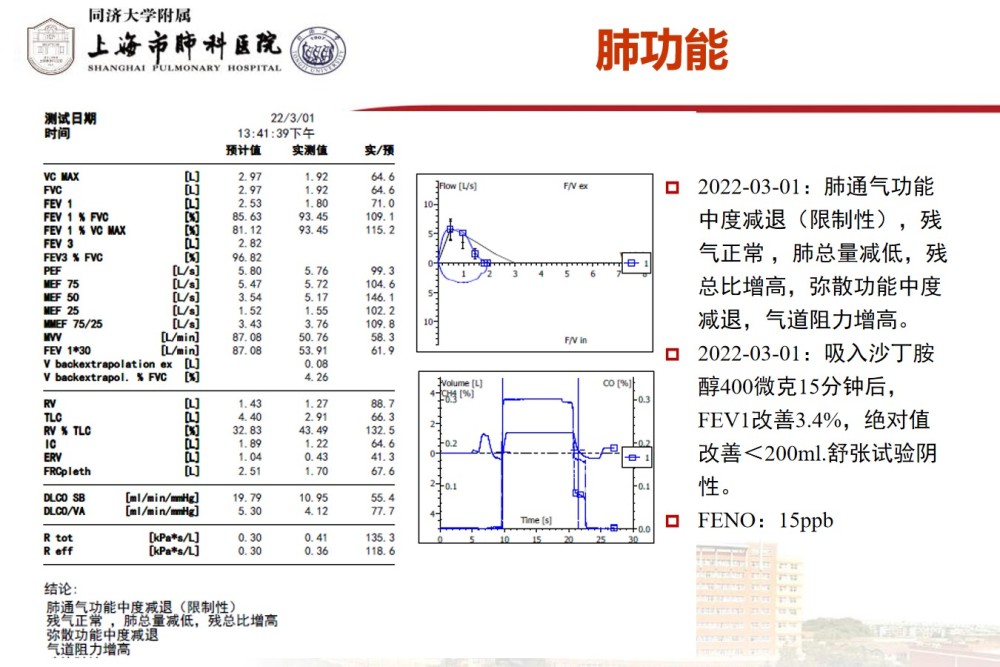
Pulmonary function showed: moderate decrease in ventilation function (restrictive), moderate decrease in diffusing function.
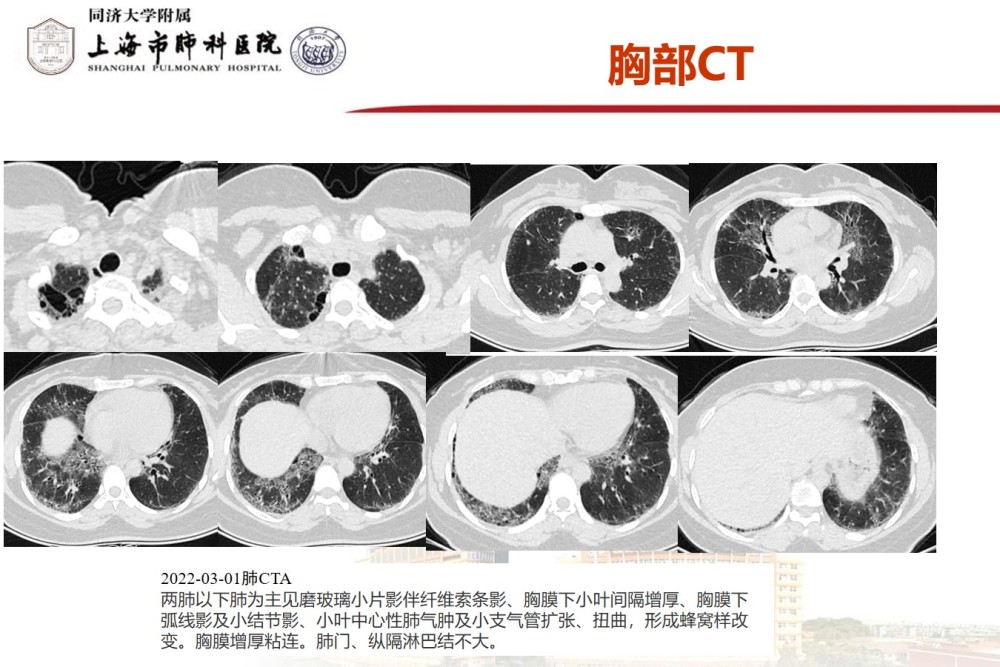
Our hospital inspection report:
Blood gas analysis: oxygen partial pressure 79.3mmHg, oxygen saturation 95.9%, alveolar arterial oxygen partial pressure 26.6mmHg
Blood count: normal
Blood coagulation: normal.
Myocardial quadruple: myoglobin 22.9ng/ml
Biochemical: glucose 3.7mmol/L, lactate dehydrogenase 268IU/L.
Blood lipids: total cholesterol (TCH) 5.63mmol/L, low density lipoprotein (LDL-C) 3.79mmol/L, high density lipoprotein (HDL-C) 0.91mmol/L< /p>
Angiotensin Converting Enzyme (SACE) 71IU/L
erythrocyte sedimentation rate (ESR) 50mm/h
Immunoglobulin: Immunoglobulin G (IGG) 17.20g/L, Immunoglobulin A (IGA) 4.28g/L
Nine joint tests: negative.
Hepatitis B Surface Antigen (HBSAG-C)>165.00ng/ml
Mycobacterium tuberculosis T-cell immune response (QFT): negative
Tumor markers: CYFRA21-1 (CA211) 3.59ng/ml, ferritin (SF) 148.75ng/ml
Sputum tuberculosis, bacterial, fungal smear and culture: Negative.
GM test, G test, Cryptococcus antigen: negative.
Inspection Report:
2022-03-01 Ultrasound of lower extremity veins: no obvious thrombosis in the deep veins of bilateral lower extremities
2022-03-02 Echocardiography: the size of each atrioventricular cavity was normal, and the left ventricular systolic and diastolic function was normal
2022-03-02 Color Doppler Ultrasound of Thyroid: Cystic Nodule in Right Lobe of Thyroid (Large 1.8×1.0mm) Proposed TI-RADS Category 1
2022-03-01 Abdominal ultrasound: normal
2022-02-28 ECG report examination report: 1, sinus rhythm 2, low voltage of limb leads 3, left axis deviation
Bronchoscopic examination:
Pathology results:
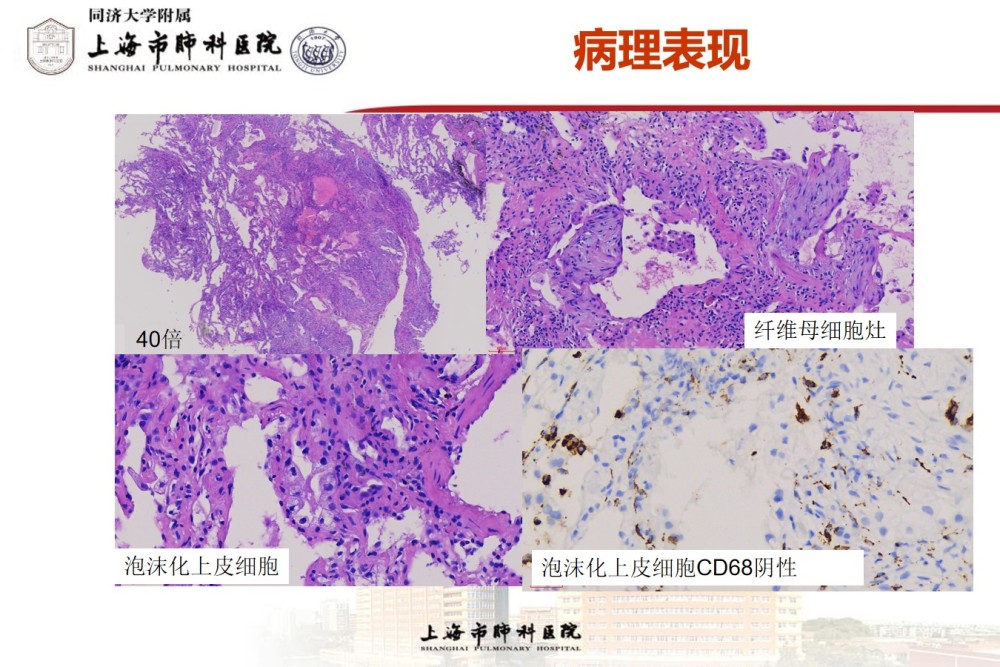

Pathological diagnosis:
(right lower basal cryo-lung biopsy) alveolar septum widening and fibrosis, focal fibrous foci in some areas, alveolar epithelial cell hyperplasia, cytoplasmic translucent and foamy change, combined with the patient’s eye and hair albinism, it is considered to be Hermansky-Pudlak syndrome-induced interstitial pneumonia, and it is recommended to further combine clinical and related genetic testing. Immunohistochemical results: TTF-1 (SPT24) (+), CD68 (histiocytes +), CK (+).
HPS is one of the albinism syndromes, which is autosomal recessive and has obvious genetic heterogeneity. First reported by Hermanky and pudlak in 1959. Clinically, it is mainly characterized by the triad of oculocutaneous albinism-like symptoms, bleeding tendency and accumulation of ceroid lipids in tissues, which may be accompanied by fatal complications such as pulmonary fibrosis, granulomatous colitis, renal failure and cardiomyopathy. .
The prevalence of HPS is estimated to be 1-9 per 1 million people.
In humans, 8 genotypes of HPS have been identified, caused by mutations in 8 different genes, namely HPS1, ADTB3A, HPS3, HPS4, HPS5, HPS6, DTNBP1 and BLOC1S3 , of which HPS1 is the most common, accounting for about half of all cases.
Encodes lysosomes or lysosome-related organelles (including melanosomes, dense platelet granules, and lamellar bodies)
The most prominent clinical manifestations of HPS are albinism due to loss of skin and iris pigment and bleeding tendency due to abnormal platelet function.
HPS pulmonary fibrosis (HPS-PF) occurs in patients with the HPS-1, HPS-2, and HPS-4 subtypes. Onset usually occurs between the ages of 30-40 or 50-60 years, depending on the individual’s genetic makeup and response to inflammation. Much like IPF, HPS-PF causes progressive and irreversible scarring of lung tissue, ultimately leading to respiratory failure and death within about 10 years after the onset of HPS-PF.
HPS1 is one of the most severe subtypes of HPS, often associated with fatal pulmonary fibrosis, leading to death.
HPS-PF has a similar histological pattern to idiopathic pulmonary fibrosis (IPF), characterized by dyspnea and progressive hypoxemia.
Treatment
pirfenidone: A pilot trial investigated the drug’s efficacy in people with mild to moderate HPS-PF. However, the study was terminated for futility. (Gahlet al., 2002)
Nintedanib
Gene therapy and gene editing are potential treatments for HPS. A preclinical proof-of-principle study demonstrated that HPS1 deficiency in skin melanocytes from HPS-1 patients could be corrected by transduction of cells with a lentiviral vector containing the HPS1 construct.
Lung transplantation remains the only treatment available for patients with HPS-PF. Patients with HPS-PF should be evaluated for lung transplantation at an early stage of the disease. Bleeding diathesis associated with HPS is not a major barrier to surgery in HPS patients, although it may be a potential contraindication because platelet dense body deficiency can lead to bleeding. Despite the bleeding risk, patients with HPS-1 underwent successful lung transplantation (El Chemaly and Young, 2016).
HPS patients with pulmonary fibrosis – namely HPS-1, -4 and HPS-2 should be evaluated for lung transplantation early in the disease process.
The disease is still an extremely rare disease. A total of 14 cases have been reported worldwide, of which 13 are foreign cases, and one is a case in my country. This case we diagnosed is the latest An example found.