One. General description
Williams syndrome (Williams-Beuren syndrome, WS), also known as Williams-Beuren syndrome, is a rare genetic disorder. It is characterized by perinatal growth retardation (prenatal and postnatal growth retardation), short stature, varying degrees of intellectual disability, and distinctive facial features that usually become more pronounced with age.
However, this group is friendly, outgoing, and talkative; therefore, it is often referred to as the “socialite disease” because they like to socialize.
II. Clinical manifestations
Williams syndrome is characterized by a wide range of symptoms and physical characteristics that vary widely in extent and severity, even among affected family members. People with Williams syndrome do not experience all of the symptoms listed below. Some affected individuals do not have heart abnormalities; others may not have elevated calcium levels (hypercalcemia). Additionally, the severity of these symptoms tends to vary from case to case.
1
Overall nutritional status
Some children with Williams syndrome may have low birth weight, poor feeding, and poor weight gain and growth (stunting). Symptoms such as vomiting, vomiting, diarrhea, and constipation are common in infancy.
Some affected infants may have elevated calcium levels in the blood (hypercalcemia), resulting in loss of appetite, irritability, confusion, weakness, fatigue, and/or abdominal and muscle pain. Calcium levels usually return to normal around 12 months.
However, in some cases, hypercalcemia may persist into adulthood. Linear growth may be delayed during the first four years of life. However, growth spurts usually occur between the ages of 5 and 10. Most people with Williams syndrome are below average in adulthood.
2
Facial Features
Newborns with Williams syndrome have characteristic “pixie-like” facial features, including unusually small heads (microcephaly), plump Cheeks, unusually broad forehead, puffiness around eyes and lips, sunken nose bridge, wide nose and/or unusually wide and prominent open mouth.
Other features may include vertical skin folds at the inner corners of the eyes (medial canthal folds), a small pointed chin, prominent ears, and/or an abnormally long vertical groove in the center of the upper lip (in human). Some babies with Williams syndrome may have dental abnormalities, including misshapen teeth (ie, hypoplastic enamel), small teeth (small teeth), and upper and lower teeth that don’t fit together properly (malbite). Commonly known as elf face
3
Eye Features
About 50% of children with this disorder may develop a stellate (star) pattern in the iris of the eye. It is most pronounced in blue- or green-eyed infants. In those with darker eyes, the pattern may be more difficult to see, or may not be present. Affected infants may also have eyes that turn inward (esotropia) and farsightedness (hyperopia).
4
Hearing
Children with Williams syndrome are extremely sensitive to sounds and may overreact to unusually loud or high-pitched sounds (hyperacusis). A chronic middle ear infection (otitis media) is often present.
5
Motor Skills
Motor development (eg, sitting and walking) and/or gross and fine motor skills (eg, picking up objects) may be delayed. Children with this disorder may experience premature development of secondary sexual characteristics such as pubic and armpit hair (precocious puberty). Breast development and menstruation may occur earlier than expected in women with Williams syndrome. People with this disorder may also have an unusually hoarse voice.
6
Cardiovascular
About 75% of children with Williams syndrome develop a congenital heart defect (CHD). The most common defect is supravalvular aortic stenosis, which is characterized by narrowing of the aorta above the aortic valve. The aorta is the main artery of the vascular system. Blood enters the aorta from the left ventricle of the heart through the aortic valve.
In supravalvular aortic stenosis, the area above the aortic valve becomes abnormally narrowed. Symptoms may include fatigue, chest pain, dizziness, abnormal heart sounds (murmur), and/or temporary loss of consciousness (syncope). The degree of aortic narrowing may vary among affected individuals.
Other congenital heart defects associated with Williams syndrome may include pulmonary stenosis and/or septal defects. (For more information on these heart defects, see the related disorders section of this report.) Unusually high blood pressure (hypertension) is also common in adults with this disorder.
7
Mental development
Children with Williams syndrome are generally friendly, outgoing, and talkative. Appropriate use of language and vocabulary range may be abnormally enhanced in some children with this disorder. Mild to moderate mental retardation may occur.
However, some children have average intelligence and severe learning disabilities. Although most affected individuals have good long-term memory, hyperactivity and attention deficit disorders are also common. Some affected individuals may have visual impairment; heThey may tend to look at the parts of the picture rather than the whole.
8
Bone development
Older children and adults with Williams syndrome may develop progressive joint problems that limit their range of motion. Skeletal abnormalities, such as curvature of the spine backwards (lordosis), front to back (kyphosis), and left to right (scoliosis), may also be present.
Some affected individuals may have a sunken sternum (pectus excavatum) and a turning of the big toe inward toward the other toes (hallum valgus). Abnormalities in the bones and joints may cause abnormal walking patterns (gait instability). Skeletal abnormalities may get worse as affected individuals age.
9
Other Developments
Some people with Williams syndrome may have other abnormalities, includingkidney (kidney) abnormalities, chronic urinary tract infections, hypoplastic (hypoplastic) thyroid glands, and umbilical or groin hernias hernia.
Three, the cause
Most cases of Williams syndrome appear to occur spontaneously (occasionally) for unknown reasons. However, some familial cases of the disease have also been reported. Ongoing research suggests that sporadic and familial Williams syndrome results from the loss of genetic material in adjacent genes (contiguous genes) located on the long arm (q) of chromosome 7 (7q11.23). This chromosomal region has been named “Williams-Beuren syndrome chromosome region 1”.
Chromosomes that reside in the nucleus of human cells carry each person’s genetic information. Human chromosome pairs are numbered from 1 to 22, along with a 23rd pair of sex chromosomes, including one X and one Y chromosome in males and two X chromosomes in females. Each chromosome has a short arm, called “p,” and a long arm, called “q.”
The chromosomes are further subdivided into numbered bands. For example, “chromosome 11p13” refers to band 13 on the short arm of chromosome 11. The numbered bands specify the location of thousands of genes present on each chromosome.
Twenty-eight genes within the 7q11.23 chromosomal region may play a causative role in Williams syndrome, including those called ELN (elastin) genes, LIMK1 (or LIM kinase-1) gene and RFC2 (replication factor C, subunit 2) gene. The LIMK1 gene is thought to be involved in visuospatial problems associated with Williams syndrome.
In familial cases, Williams syndrome is inherited as an autosomal dominant trait. Genetic diseases are determined by two genes, one from the father and one from the mother. A dominantly inherited disorder occurs when only a single copy of an abnormal gene is required for the disease to appear. The abnormal gene can be inherited from either parent, or it can be the result of a new mutation (genetic change) in the affected individual. Regardless of the sex of the child, each pregnancy has a 50% risk of passing the abnormal gene from an affected parent to the offspring.
Hypercalcemia associated with Williams syndrome may occur due to abnormal sensitivity to vitamin D.
The American Academy of Pediatrics has developed a diagnostic score:

IV. Incidence
Williams syndrome is a rare disorder that affects an equal number of men and women and can affect babies of any race. In the United States, the disease affects about 1 in 10,000-20,000 births.
Williams syndrome has an incidence of 1/7 500 in Norway and 1/23 500 of live births in Hong Kong, China. There is no relevant epidemiology in mainland China. Investigation report.
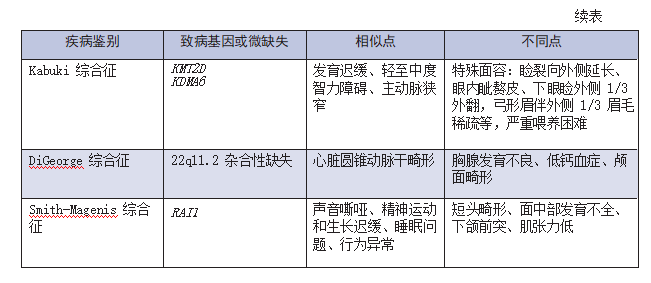
V. Differential Diagnosis
The following conditions may have symptoms similar to those of Williams syndrome. Comparison may help in differential diagnosis:
Noonan syndrome is a rare genetic disorder that is usually evident at birth (congenital). The disease can be characterized by a range of symptoms and physical features that vary widely in scope and severity. In many affected individuals, associated abnormalities include a distinctive facial appearance; a wide or webbed neck; a low hairline at the back of the head; and short stature.
Characteristic abnormalities in the head and face (craniofacial) region may include wide eye (hypertopia); vertical skin folds (medial canthal folds) that may cover the inner corner of the eye; upper Drooping eyelids (ptosis); small jaws (minignathia); low bridge of the nose; and low, prominent, abnormally rotated ears (pinnae). There are also often significant skeletal deformities, such as abnormalities of the sternum (sternum), curvature of the spine (kyphosis and/or scoliosis), and outward deviation of the elbows (cubitus valgus).
Many babies with Noonan syndrome also have heart (heart) defects, such as blockage of normal blood flow from the lower right chamber of the heart to the lungs (pulmonic stenosis). Other abnormalities may include malformations of certain blood and lymphatic vessels, coagulation and thrombocytopenia, mild mental retardation, failure of the testes to descend into the scrotum (cryptorchidism) in the first year of life in affected males, and/or other symptoms and findings .
Idiopathic infantile hypercalcemia is characterized by an unexplained increase in blood calcium levels in the newborn (idiopathic). Symptoms may include loss of appetite (anorexia), irritability, confusion, weakness, easy fatigue, and/or abdominal and muscle pain.
Some studies in the medical literature question whether idiopathic infantile hypercalcemia is a different disorder from Williams syndrome or a variant of the same disorder. Babies with this disorder do not have the characteristic facial features or heart associated with Williams syndromedefect.
Leprechaunism is a rare, progressive, inherited endocrine disorder. It is characterized by overgrowth (hyperplasia) of the pancreas, inability to use insulin properly (insulin resistance), and excess estrogen. Growth retardation begins during fetal development.
Symptoms of goblinism may include short arms and legs, large hands, a pixie-like face, sunken cheeks, a pointed jaw, a wide flat nose, low-set ears, and wide eyes. Children with goblin disorder often have low levels of circulating glucose (hypoglycemia) and elevated insulin levels (hyperinsulinemia). People with this condition cannot use insulin effectively.
6. Diagnosis
The diagnosis of Williams syndrome can be confirmed with a thorough clinical evaluation, including a detailed patient history and specialized blood tests that can detect elevated calcium levels in the blood. Another test called fluorescence in situ hybridization [FISH] can be used to determine if there is a deletion of one of the elastin genes on chromosome 7. This deletion is thought to occur in most people with Williams syndrome.
Diagnostic Process
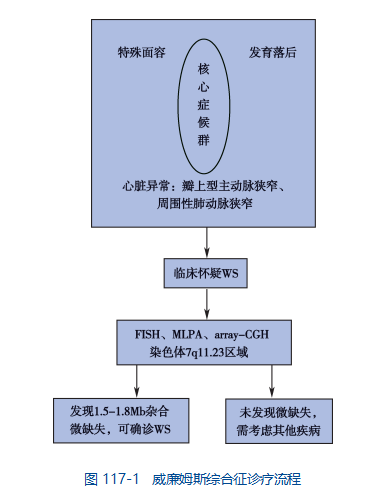
Seven, standard treatment
1
Treatment
(1) Infants with Williams syndrome with elevated blood calcium levels may be placed on a vitamin D-restricted diet. Calcium intake may also be limited. For those children with severe hypercalcemia, treatment with corticosteroids such as prednisone may be temporarily considered. Calcium levels usually return to normal after about 12 months of age, even in untreated babies.
(2) Affected children with symptoms related to heart defects should undergo a thorough evaluation at a hospital familiar with these rare congenital heart diseases. Specialized tests can be performed to determine the severity and exact location of congenital heart defects (ie, ECG, echocardiography, or cardiac catheterization). Some children with Williams syndrome with severe heart defects may need surgery to repair the defect.
(3) Special education services at centers and schools for children with developmental disabilities may benefit children with Williams syndrome in reaching their individual potential. A supportive team approach may also be helpful. Includes speech and language therapy, occupational and physical therapy, social services and/or vocational training. Music therapy has long been advocated, but thought to be unproven, because it enhances learning and relieves anxiety in people with Williams syndrome.
(4) Genetic counseling may benefit people with Williams syndrome and their families. Other treatments are symptomatic and supportive.
2
Rehabilitation
(1) Sensory integration training: Vestibular (including gravity and movement), proprioception (including muscle and sensation), and touch and other multi-sensory stimulation of the whole body movement, During training, children are given various stimuli such as vestibular, muscle, joint, skin touch, sight, hearing, smell, etc., and combine these stimuli with exercise.
(2) Muscle strength and core stability training: Through sitting, four-point support, kneeling, squatting, standing and other anti-gravity training and resistance Resistance training activates the core muscles of the child, thereby enhancing the quality of their posture.

Image source: Photo Network
(3) Game therapy: Through games that interest children, let children actively accept language, movement, communication, cognition and behavioral training.
(4) Activity observation training: Let children actively observe people (smile, tongue out, nod and facial expression changes, etc.) or objects (toys, personalized and special equipment) for repeated active imitation training.
(5) Correct cognitive and behavioral problems through psychological and cognitive behavioral therapy.
VIII. Prognosis
Intrinsic can involve multiple systems such as cardiovascular and endocrine, and the prognosis depends on the function of each organ in the affected system. If the organ damage is not severe, the prognosis is relatively good; severe organ damage, especially severe cardiovascular damage, can affect survival, and children often die due to heart failure or sudden death. Complications such as hypertension can also accelerate the development of cardiovascular disease in adulthood and affect prognosis.
By Mays Medicine
Edit: Rita is for doctors to learn and communicate only