▎WuXi AppTec Content Team Editor
This Issue
[1] Dr. Richard Pazdur, director of the FDA Oncology Center of Excellence, and other experts published an article in the New England Journal of Medicine, emphasizing that the reform of accelerated approval programs requires a comprehensive strategy
[2]The European Medicines Council plans to publish guidelines for the development and manufacture of synthetic peptides and synthetic oligonucleotides
Multiple FDA experts suggest reform of accelerated approval program requires comprehensive strategy
Accelerated approval (accelerated approval) is a pathway established by the US FDA in 1992 to accelerate the development of drugs for the treatment of serious and life-threatening diseases. This pathway could lead to accelerated approval of drugs based on surrogate endpoints that predict clinical benefit, or interim clinical endpoints.
In recent years, the relationship between accelerated approval endpoints and clinical benefit, the time required to complete confirmatory clinical trials, and withdrawal of approval if confirmatory clinical trials fail to show clinical benefit The process of the disease has received extensive attention. A few days ago, a number of FDA experts published an article in the New England Journal of Medicine, pointing out that the reform of the accelerated approval program should not only focus on the process after the accelerated approval is granted, but also on the process before the accelerated approval is granted. Factors such as clinical trial design, clinical endpoints, and study patient populations that support accelerated approval and confirmation of clinical benefit should be comprehensively considered.
Status of accelerated approvals
Currently, accelerated approvals are primarily used in oncology, with cancer therapies accounting for 85% of accelerated approvals over the past 10 years. In these accelerated approval indications, 50% of the drug’s clinical benefit was confirmed by subsequent clinical data. 12% of indications were withdrawn. The median time to validation of clinical benefit and conversion of accelerated approval to full approval was 3.1 years. The median time to withdrawal was 3.8 years.
The time required to complete a confirmatory clinical trial and the time required to withdraw an indication after a confirmatory clinical trial has not shown clinical benefit are of widespread concern. In this regard, FDA experts pointed out that one of the key factors in the length of time required is whether the drug developer has already initiated confirmatory clinical trials when the accelerated approval is obtained.
For drugs that have already started confirmatory clinical trials at the time of accelerated approval, either because of the conversion of accelerated approval to full approval because of clinical benefit verification, or because of no clinical benefit demonstration For withdrawal of indications, the time required for withdrawal was relatively short (median time was 3.0 years and 3.8 years, respectively). And if the developer had not initiated confirmatory clinical trials at the time of accelerated approval, the median time to full approval was extended to 5.1 years, and the median withdrawal of indications due to lack of clinical benefit validation Time to reach 7.3 years.
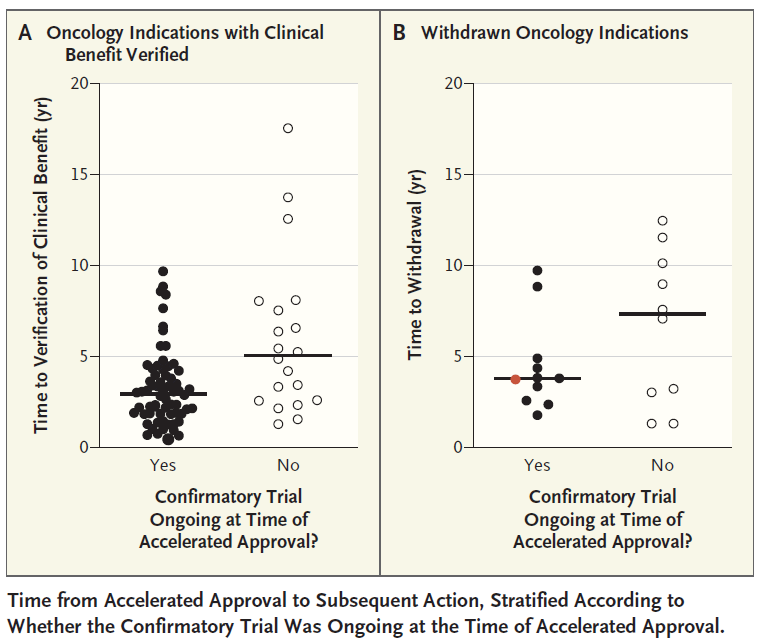
▲Time from Accelerated Approval to Next Regulatory Action (Image Source: Reference [1])
FDA experts say that although these areas have received more attention, reforming the accelerated approval program should also focus on the development of drug developers in consultation with the FDA before obtaining accelerated approval. A plan for confirming clinical benefit, reducing the time required to confirm clinical benefit.
Consideration of Surrogate Clinical Endpoints
For example, in terms of clinical endpoints, the most common surrogate endpoint used in oncology to support accelerated approval is durable overall response rate (ORR). ORR is a reliable marker of drug activity, as malignant tumors usually do not shrink spontaneously without treatment. However, the relationship between ORR and overall survival is uncertain across cancer types and drug types. For example, immune checkpoint inhibitors represented by anti-PD-1/PD-L1 antibodies may show significant overall survival benefits, but they have no great effect on ORR indicators.
Clinical Trial Design Considerations
In terms of clinical trial design, FDA expert opinion is: Drug developers may consider directly initiating a randomized clinical trial to support both accelerated approval and subsequent validation of clinical benefit . Accelerated approval can be based on a preplanned analysis of the overall response rate, while full approval can be based on clinical benefit (usually an overall survival benefit) demonstrated after the clinical trial is fully concluded.
This strategy can provide a more comprehensive safety assessment and more definitive evidence to support the benefit/risk ratio assessment. It also reduces the risk of prematurely terminating programs that show limited improvement in overall response rates but prolong overall survival. And this clinical trial could consider enrolling patients with relatively mild disease severity, potentially reaching a wider patient population if the effect is positive.
Another strategy is to conduct two studies at the same time: a single-arm study to support accelerated approval based on overall response rate to recruit patients with limited treatment options; another randomized study As a confirmatory study, patients who received less prior treatment were recruited. If the two trials are started at the same time, the safety data and interim overall response rate from the confirmatory study could provide more supporting evidence and increase confidence in granting accelerated approval.
Related Links:
https:https://www.nejm.org/doi/full/10.1056/NEJMp2208954
European Medicines Agency plans to develop guidelines for the development and manufacture of synthetic peptides and synthetic oligonucleotides
Recently, the European Medicines Agency (EMA) issued a document stating that given the manufacturing process, analysis, characterization and quality standards of synthetic peptides and synthetic oligonucleotides, there are still many aspects that have not been The EMA plans to issue guidelines for the development and manufacture of synthetic peptides and synthetic oligonucleotides.
Related Links:
https:https://www.ema.europa.eu/en/establishment-guideline-development-manufacture-synthetic-oligonucleotides
https:https://www.ema.europa.eu/en/establishment-guideline-development-manufacture-synthetic-peptides
WuXi AppTec provides integrated, end-to-end new drug R&D and production services for the global biopharmaceutical industry, covering chemical drug R&D and production, biological research, preclinical testing and clinical trials R&D, cell and gene therapy R&D, testing and production. If you have relevant business needs, please click the picture below to fill in the specific information.